

> Abstract
Traumatic brain injury (TBI) is a frequent occurrence in dogs and cats and it is mainly caused by motor vehicle accident, fall, human violent acts and attacks from other animals. Damages in TBI are divided in primary and secondary. Primary damages take place immediately as a result of the direct mechanical destruction of the neural tissue at the time of trauma, while secondary brain damages occur within a few minutes or days following the traumatic event and they are caused by systemic extracranial injuries and intracranial biochemical alterations.
Initial assessment of an animal with TBI is focused on the life-threatening injuries and it is followed by the performance of neurologic examination.
It is difficult for the clinician to control primary brain damage. Treatment efforts must start immediately and their aim is to stabilize the animal, prevent and treat the secondary brain damages. At first general measures are taken to restore and maintain brain oxygenation. This is achieved by supporting the circulatory system with fluids and by oxygen supplementation. The aim of instituting specific measures is to minimize the brain injury.
TBI is associated with high mortality rates in both humans and animals. However, dogs and cats exhibit remarkable rehabilitation ability, provided that extended follow up is granted after a severe brain trauma. For this reason, it is strongly recommended to not infer hasty conclusions about prognosis based on the initial status of an animal presented with TBI.
> Introduction
Severe traumatic brain injury (TBI) is associated with high morbidity rates in humans and animals1 and it may result from motor vehicle accident, fall, inadvertent or malicious human violent acts, attacks and bites from other animals and missile2. In a multicenter survey, 26% of the traumatized dogs and 42% of the traumatized cats manifested TBI3. In another study, including 122 cases of TBI, most of the dogs were admitted after motor vehicle accident4.
The appropriate treatment of TBI is subject of continuous research in neurosurgery and intensive care in both human and veterinary medicine. However, in small animal practice due to lack of evidence from retrospective or prospective studies, most of the guidelines are based on experimental studies, clinical studies in humans and personal experience.1 It is widely accepted though that immediate and aggressive treatment may increase survival rates. After the initial treatment of life-threatening injuries, like open pneumothorax, airways obstruction etc., clinician’s efforts are targeted on maintaining cerebral perfusion pressure.5 Dogs and cats exhibit good rehabilitation ability, even after substantial brain tissue loss, provided that extended follow up is granted after a major brain trauma.
> Pathophysiology
TBI causes primary and secondary brain damages. Primary brain damages occur immediately as the consequence of mechanical impact to the neural tissues at the time of trauma and they ignite complex inflammatory processes which end up to secondary brain damages. The latter damages occur minutes to days following the trauma and they are caused by a combination of systemic extracranial injuries and intracranial biochemical and physiological alterations.2
Primary brain damages resulting in natural disorganization of intracranial structures are the following:
1. contusion
2. axonotmesis (axon injury)
3. mechanical rupture by immersion of the fracture fragment (depressed fracture)
4. hematoma
The mildest injury which is characterized by transient consciousness loss is called concussion. Albeit this kind of injury is not accompanied by histological changes of the neural tissue, posttraumatic clinical signs are variable.5
The most severe primary brain damage is the mechanical rupture of brain parenchyma. Direct damage to the vessels may lead to intracranial hemorrhage and/or vasogenic edema. Hematomas are distinguished in axonal located in brain parenchyma and peripheral ones which are located in the subarachnoid, subscleral and episcleral space. Peripheral hematomas may lead to brain compression and serious neurologic disturbance. 6 Previously, it was believed that these hematomas are rare in dogs and cats suffering from TBI. However, in recent studies it has been demonstrated that peripheral hematomas occur in 10% of animals with mild TBI and in over 80% of animals with severe TBI.7 Treatment of primary brain damages is beyond the clinician s abilities and his main concern should be the prevention, recognition and treatment of secondary brain damages.8
Primary brain damage triggers a large number of interdependent biochemical reactions which further deteriorate neural tissue function and lead to cell death. Those biochemical reactions constitute what is called secondary damage. Secondary damage is mainly mediated through increased release of excitatory neurotransmitters, depletion of energy stores and production of free oxygen radicals and post inflammatory cytokines, which all result in neuronal injury. Intracellular increase in sodium and calcium ions concentration, extracellular increase in potassium ions concentration, accumulation of lactate, disturbance in the balance between thrombosis and fibrinolysis and activation of complement are also involved. Possible consequences following secondary neural tissue damage are formation of edema, increased intracranial pressure, changes in the integrity of blood-brain barrier and alterations in cardiovascular function.1,8,9
Apart from the above mentioned intracranial damages, systemic consequences of the initial trauma may also contribute to secondary brain damage. These include hypotension, hypoxia, hyper- or hypoglycemia, hyper- or hypocapnia and hypothermia.1,5 Those extracranial alterations may worsen brain trauma due to compromised cerebral perfusion and can even lead to brain tissue herniation (e.g. herniation of the cerebellum through the occipital foramen).10
Intracranial pressure
In the traumatized area of the brain, contrary to metabolic activity which remains constant or increases, blood flow decreases and the supply of oxygen, glucose and ATP molecules is diminished. Thus, ion pumps of the cellular membranes are rendered underactive which causes accumulation of sodium and calcium ions (intracellulary) and potassium ions (extracellulary) and ultimately cytotoxic edema. It is therefore clear the significance of cerebral blood flow (CBF) preservation.11
Cerebral blood flow (CBF) depends on the cerebral perfusion pressure (CPP) and the cerebral vascular resistance (CVR) and it is defined as:
CBF = CPP / CVR11
Regional compensatory mechanisms regulate CVR, and through them CPP is remained stable when mean arterial pressure (MAP) fluctuates from 50 to 150 mmHg. Auto regulatory mechanisms are influenced by the acid-base balance of the neural tissue, the adequate supply of oxygen and the subsequent removal of CO2 . When auto regulatory mechanisms are disturbed-which is the case in TBI-, CBF depends further more on CPP. Consequently, even minor reduction in CPP can provoke changes in CBF and lead to ischemia of the brain parenchyma.2,9,11
Cerebral perfusion pressure is the force which drives the blood to the brain, providing oxygen and necessary nutrients to its parenchyma. It is also the main factor which determines CBF and it is defined as the difference between MAP and intracranial pressure (IP):
CPP = MAP – IP11
Increased intracranial pressure is a common and potential fatal sequel of TBI. Intracranial pressure is the pressure that prevails in the calvarium and it is exert by the brain tissue, the arterial and venous blood and the cerebrospinal fluid (CF). Under normal circumstances it ranges from 5 to 12 mmHg. When the volume of one of the abovementioned anatomical elements is increased, the volume of one or both of the other two elements compensatory has to decrease if intracranial pressure is to remain stable. This property is known as intracranial compliance. Intracranial compliance decreases when IP increases. When the latter increases beyond the limits that are settled by the effectiveness of the compensatory mechanisms, cerebral perfusion is lessened and cerebral ischemia turns up.9-13
In TBI, due to the fact that dimensions of the calvarium are fixed, when the intracranial volume increases by edema or hemorrhage, intracranial pressure increases. Serious elevations in intracranial pressure stimulate the ischemic reflex of the brain or Cushing’s reflex. In particular, diminished blood flow to the brain causes increase in CO2 concentration, which is detected by the vasomotor center. The latter stimulates the sympathetic nervous system which in turn increases the MAP, as a means of maintaining the CPP. Systemic hypertension is detected by baroceptors in the carotidic body and aortic arch, resulting in compensatory bradycardia. Ischemic reflex is a late occurrence and probably dictates life-threatening increase of intracranial pressure which calls for aggressive treatment.14,15
In cases of MAP reduction (systemic hypotension), CPP also decreases resulting in inadequate blood supply to the brain and hypoxia of the neural tissue. Vasodilation of the brain vessels during hypotension is not sufficient for CPP maintenance. Furthermore, secondary autolysis of the neural tissue is exacerbated by hypotension and hypoxia ending up in further damage of the nervous tissue, edema and raised intracranial pressure. 15,16
In people with TBI, episodes of hyperglycemia are often encountered and it is assumed that they are associated with poor prognosis.17 Aerobic metabolism of glucose provides energy to the brain. Disturbance of brain metabolism combined with the increased requirements in energy derived from glucose in patients suffering from TBI, lead to reduced energy supply.18 Moreover, concurrent activation of sympathetic nervous system and increased release of catecholamines result in raised intracranial pressure, increased demands in oxygen and increase in the levels of glucose in the blood.19 Hyperglycemia, which is an indication of stress response, is associated with the severity of injury in dogs and cats, but it has not been linked with prognosis.4 Therefore blood glucose measurement is recommended in animals with TBI, albeit in humans glucose level in peripheral blood does not exactly match with that of the brain.17
> Assessment of animal with TBI
Assessment of the status of an animal presented with TBI, starts by addressing the life threatening injuries. Studies have shown that approximately 60 % of people with TBI suffer from concurrent injuries20 and 25 % of these injuries are related to intrathoracic or intraabdominal organs. As in every trauma patient, initially the animal is scrutinized and treated for injuries concerning the airways, the respiratory function and the blood circulation. Hypovolemia and hypoxia are associated with increased mortality in people with TBI1,14 and thus clinician must be able to recognize and treat them immediately. At the same time, continuous monitoring of the vital signs prevents their occurrence. 8
Neurological status of an animal with TBI is often not stable thus serial physical examinations are recommended. The initial neurological examination includes assessment of level of consciousness, size of the pupil and its response to light, position and movement of the eyeballs and movement of the limbs.5
In humans TBI is characterized as mild, medium or severe according to Glasgow Coma Scale (GCS). In veterinary medicine, a modified Glasgow Coma Scale (MGCS) is being used and it is based on the following assessments:
- level of consciousness
- motor activity (including position of the body)
- activity of the brainstem (pupil size and its response to light, activity of the vestibular system) 8
Each of the three assessment classes is scored from 1 (worst clinical sign) to 6 (normal). Total score can range from 3 to 18. When the score is higher the prognosis is better. In veterinary medicine the prognostic significance of the assessments based on MGCS has been tested in relation to survival rates at the first 48 hours after the injury. 21 Total score in each moment reflects the severity of the underlying brain injury, its progress and the results from treatment efforts. In people, if the total score varies from 3 to 9 during the first 24 hours, scoring is linearly linked with bad prognosis. 22 However, these retrospective patterns of prognosis may not be as useful as it seems and at the moment prospective studies are lacking.21 It is thus suggested that MGCS can be utilized for the evaluation of clinical signs amelioration.
Level of consciousness is the most reliable empirical means of assessing the impairment of brain function after TBI and provides information about the functional ability of the cortex and the activating reticular system. If the injury involves these structures, the patient is in coma or stupor, and the prognosis is guarded.10,23
Motor activity is controlled by the red nucleus located in the mesencephalus and it is influenced by the animal’s level of consciousness. Abnormal posture like opisthotonus and hyperextension of the limbs can be observed in cases of disconnection of the brain stem and the telencephalus (decerebrate rigidity) and it has to be differentiated from cerebellar rigidity and Schiff-Sherrington syndrome. (Figures 1, 2 and 3). The main difference between cerebral and cerebellum injury is the alteration in the level of consciousness because in cerebellar rigidity the level of consciousness is spared while injury of the mesencephalus leads to coma with bad prognosis.21,23 In localized injuries, motor disorder concerns the contralateral part of the body. If the lesion involves the brainstem or the cortex, postural reaction tests are weak ipsilateral or contralateral, respectively.24

Normal function of the brainstem is ascertained by performing neuro-ophthalmic examination. Cranial nerves and size of the pupil and its response to light are being examined. Normal reaction of the pupil indicates good function of the rostral part of brainstem, optic chiasm, optic nerves and retina. Mydriasis may suggest herniation of brain hemispheres, while fixed and medium sized pupils which do not respond to light stimuli are observed in cases of cerebellum herniation. Menace reflex is useful for the neuroanatomical localization of injury but it is without prognostic significance. However, the function of the vestibular system, which clinically is assessed by the presence of normal or pathological nystagmus, is considered to be prognostic.10,14,25
Assessment of respiratory movements, albeit it is not included in MGCS, contributes to the neuroanatomical localization and the evaluation of the severity of injury. Malfunction of the respiratory system may accompany TBI, because among the concurrent injuries, pneumothorax, hemothorax, rib fractures and upper airway and pulmonary contusions are commonly reported. Function of the respiratory system is important in TBI, because it influences the metabolism of the nervous system.14 One possible and severe respiratory disorder is neurogenic pulmonary edema (NPE), which is attributed to the stimulation of the sympathetic system and the increased release of catecholamines. NPE may be detrimental for the patient; however it is usually self-limiting in a few hours or days.26
Moreover, it is recommended to monitor hematocrit and the concentration of total solids, glucose and electrolytes in peripheral blood, with special emphasis on the levels of serum glucose, because, as it was mentioned above, it seems that hyperglycemia is associated with the severity of injury in both humans and animals.4,27,28
Imaging of the skull is commonly indicated, especially in animals which do not respond or deteriorate despite their initial improvement after aggressive treatment. Plain radiographs are not particularly useful but they may reveal skull fractures. Computed tomography (CT) is preferred over magnetic resonance imaging (MRI) at the beginning of the imaging study because it provides faster and better imaging of both the bones and the location and extent of the hemorrhage. In contrast, MRI is more useful when it is performed after the initial stabilization of the animal, because it assists on the determination of prognosis for the rehabilitation of the animal. It is noted that MRI is more expensive imaging modality compared to CT.5,24
> Treatment of traumatic brain injury
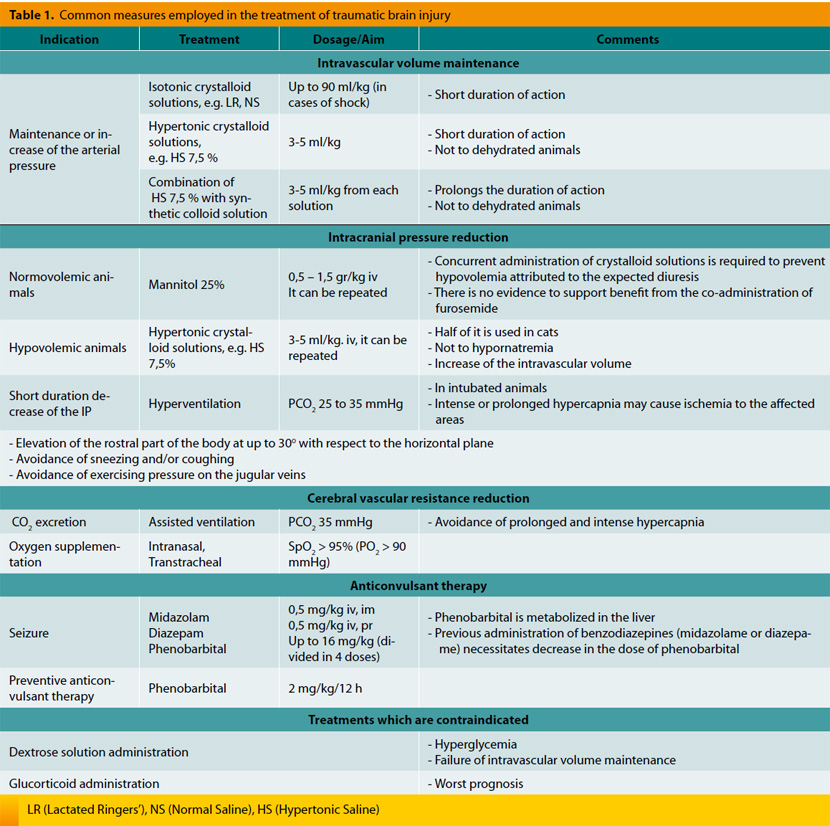
The aim of treatment in cases of TBI is the preservation of arterial blood supply to the nervous system and its protection from secondary changes (Table 1). This is accomplished with the following:
1. Preservation of perfusion
As it was previously mentioned, maintenance of adequate cerebral blood flow requires on one hand sufficient cerebral perfusion pressure and on the other hand relatively small cerebral vascular resistance.13
Maintenance of sufficient cerebral perfusion pressure
This is accomplished by treating hypovolemia and trying to maintain sufficient MAP via administration of fluids. In the past it was believed that aggressive intravenous administration of fluids may exacerbate cerebral edema. However, there are no data to support this view and thus there is no contraindication in the administration of fluids. 29 Moreover, hypotension has been associated with increased morbidity and mortality rates in humans. In a retrospective study, hypotension in cases of TBI was correlated with 150 % increase in mortality.16 It is likely that in animals presented with TBI, hypovolemia must be avoided and intravascular volume must be restored so that mean arterial pressure will remain normal.
There is no consensus on the appropriate type of fluids in the treatment of TBI.30 Available options include isotonic crystalloid, hypertonic crystalloid and synthetic colloid solutions and blood products. Intact blood-brain barrier is permeable to water but not to ions and colloid molecules due to the tight junctions between the cells of the vessels wall.31 Studies have shown that colloids do not have considerable effect on the brain water content and the IP. However, in the traumatized brain blood-brain barrier may be disrupted locally or totally and thus it may be indiscriminately permeable to ions and colloid molecules.32 For this reason, it has been proposed that non proper choice of the treatment fluids may lead to cerebral edema. However, the benefits from CPP maintenance overweigh the potential risks.14 Isotonic crystalloid solutions are administered at a rate of up to 90 ml/kg in dogs and 60 ml/kg in cats, while synthetic colloids are administered in boluses of 10-20 ml/kg in dogs and 5 ml/kg in cats, each time. Clinically, the desirable effect is mainly perceived by the decrease in heart rate, the increase in arterial blood pressure and the improvement of pulse quality and mucus membrane color.14 Hypotonic solutions (like 5 % dextrose solution, which is isotonic but its biological behavior is hypotonic) are always avoided because they do not retain fluids in the intravascular space and thus they do not increase MAP but instead they cause diffusion of water towards the interstitial space.33
Administration of hypertonic solutions is followed by rapid increase of the blood osmolality which facilitates fluid diffusion through the capillaries from the interstitial and intracellular space to the intravascular space. The result is rapid and substantial increase of the intravascular volume which exceeds the volume of fluids that has been administered34. Hypertonic solutions are able of reversing hypovolemic shock with smaller volumes of fluids and they may be the best choice of fluid therapy in patients with TBI.34,35 Sodium chloride solutions of 15 %, 7.5 % and 3 % are currently being used and preference is given in administering the lower concentrations of 7.5 % and 3 % in dosages of 4 ml/kg and 5.3 ml/kg, respectively. However, despite the fact that response to hypertonic crystalloids is rapid, subsequent distribution of fluids in the body results in reduced duration of action, which is limited to a maximum time of 75 minutes.36 Adding a synthetic colloid solution prolongs their effect for a few hours. Simultaneous administration of hypertonic and colloid solution is more effective in restoring intravascular volume compared to administration of each solution alone. Administration of 4 ml/kg of solution comprised of hypertonic crystalloid solution 23.4 % and Hetastarch 6 %8, in a rate of 1:2, has been recommended. Restoration of the intravascular volume must be followed by continuous hydration of the animal using isotonic crystalloid solutions and not hypertonic solutions, which tend to dehydrate the tissues. In fluid treatment, maintenance water needs and the ongoing losses should be taken into consideration.8
Intracranial pressure reduction
IP reduction can be achieved by simple means. Positioning the head of the animal in a higher level compared with that of its body by tilting the examination table at 15-30º, increases blood drainage from the brain and reduces its blood volume, without causing significant alterations in its oxygenation.8 It is important to remember that neck flexion should be avoided because venous drainage is impeded. For the same reason, placement of collars or jugular catheters and venipuncture of the jugular vein are also avoided. The last procedure even if it is simple and brief, it may cause intracranial pressure increase.2
Mannitol is an osmotic diuretic which is traditionally used in cases of TBI in humans and animals. 37,38 It is considered the treatment of choice for reduction of IP and improvement of CPP.39 It is administered in dosages of 0.5-1.5 gr/kg slowly, within 15-20 minutes.40 Mannitol initially expands the volume of blood and decreases its viscosity resulting in vasoconstriction and maintenance of cerebral perfusion.8 Thus, while perfusion is maintained, IP is decreased due to the decrease of intravascular volume.41 This rheological mechanism of mannitol is considered responsible for its intense action, takes place immediately after its administration and lasts for approximately 75 minutes. Osmotic effect of mannitol starts 15-30 minutes following its administration. In healthy brain, mannitol advocates the diffusion of fluid from the interstitial and intracellular space towards the intravascular space, causing osmotic diuresis. In the past, the effect of mannitol in the traumatized brain was questioned due to the compromised perfusion of its parenchyma.40 In theory, another concern regarding mannitol’s administration is the worsening of intracranial hemorrhage from the osmotic effect exert to the extra vascular space of the traumatized nervous system. However, there is no clinical evidence to support those concerns and on the grounds that the benefits from the treatment overweigh the supposed risks, mannitol constitutes the treatment of choice in cases of cerebral edema due to TBI.1,2 Mannitol is also believed to reduce free oxygen radicals.42
Mannitol is contraindicated in hypovolemic patients because its diuretic action may worsen hypovolemia and lead to hypotension. Thus it is suggested to be administered only in normovolemic patients who receive adequate treatment with crystalloid solutions.10
Sodium chloride hypertonic solutions consist another option for the treatment of cerebral edema and increased IP due to the inability of sodium to pass through the blood-brain barrier. Those solutions exert osmotic effect and promote the shift of water from the intracellular and interstitial space towards the intravascular space, leading to decrease of the water volume in the interstitial and intracellular space of the brain and as a result decrease of the IP. Sodium chloride hypertonic solutions also improve local perfusion because they dehydrate the endothelial cells located in the wall of brain’s vessels. The onset of their action is very fast and besides IP reduction, they also stabilize circulatory system. The action regarding the increase of intravascular volume lasts for 15-75 minutes36 and that related to IP reduction, even more.43 If they are administered in large volumes and because of the rapid increase of the intravascular volume there is danger in worsening concurrent pulmonary edema or concussion. Nevertheless, the same danger applies to mannitol, too.44
Pretty enough studies which compare the action of mannitol and that of hypertonic solutions suggest that hypertonic solutions are more effective in treating increased IP.43,45 At the moment and according to guidelines in humans, mannitol is still considered the drug of choice for the treatment of TBI. However, recent studies strengthen the increased clinical interest in hypertonic solutions which might be proven more effective and with fewer side effects.46 More studies are needed to infer safe conclusions about the proper use and the indicated concentration of hypertonic solutions in the treatment of animals with TBI.
In small animal practice, contrary to human medicine, decompressive craniectomy is rarely performed for the treatment of increased IP. The procedure is indicated in cases of foreign body suspicion, hematoma formation, continuing hemorrhage, fracture, space occupying lesions and when the neurological status of the animal deteriorates despite appropriate medical treatment. 8
Cerebral vascular resistance regulation
a. Ventilation
The most important factor which determines CBF and the blood volume of the brain is partial carbon dioxide pressure in arterial blood (PaCO2) which regulates tone and diameter of brain’s vessels. Specifically, hypercapnia promotes vasodilation, decrease of the resistance in blood flow and increase of its supply, which lead to increased blood volume in the calvarium and thus IP increase. PaCO2 is measured with blood gases analysis, while in intubated patients end-expiratory CO2 is measured (ET CO2)11. Traditionally, hyperventilation of the lungs is used as a means of reducing IP. Hyperventilation causes fall in PaCO2, leading to vasoconstriction, blood volume reduction and as a sequence IP reduction. It has been found that even moderate hypocapnia (PaCO2 29-36 mmHg) results in excessive vasoconstriction and decrease of perfusion in the traumatized areas of brain which predisposes to ischemia.47 Consequently, this method must be used with caution and mainly as temporary treatment in cases of sudden IP elevation and neurological signs impairment, while PaCO2 must be maintained between 29 and 36 mmHg.47
β. Oxygenation
Oxygen deficiency is one of the most important factors which contribute to secondary brain injury. Anaerobic environment in the brain diverts metabolism to anaerobic pathways resulting in local tissue acidosis. The latter influences vascular tone in a similar manner with that of hypercapnia. Surveys in people have shown that mortality rate of patients with confirmed hypoxia is double than that of patients with normal tissue oxygenation.16 Thus oxygen supplementation is mandatory in patients with TBI. Percent saturation of hemoglobin (SpO2) should be above 95 % which corresponds to partial pressure of oxygen in arterial blood not less than 80 mmHg. If measurement of arterial blood gases is feasible, it is attempted to maintain PaO2 above 90 mmHg. Oxygen is supplemented by face mask, nasal or tracheal catheter, tracheal tube, via tracheostomy or by placing the animal in oxygen cage. Nevertheless, oxygen cage utilization is ineffective in cases of TBI, because continuing monitoring and handling of the animal does not allow for the door to remain closed for long periods, which is prerequisite in achieving adequate oxygen concentration. During oxygen therapy, the animal should not cough (e.g. intubation) or sneeze (e.g. nasal catheters) and in general, every factor which can potentially increase IP, should be avoided.1
2. Neuroprotection
Corticosteroids have been used widely in the treatment of TBI for over 30 years. The goal was the reduction of IP. Due to lack of sufficient data, a large number of researchers established a team and conducted a wide randomized prospective study (CRASH) regarding the use of corticosteroids in the treatment of TBI. It was found that in the group of patients receiving corticosteroids, death numbers were substantially increased during the first 2 weeks following the TBI. For moral reasons the study was completed sooner.48 Nowadays, it is believed that administration of corticosteroids in patients with TBI -methylprednisolone included- probably increases mortality48,49 and they are considered harmful or at least ineffective. Furthermore, corticosteroids are known to be implicated in hyperglycemia, immune suppression, delayed wound healing, gastric ulceration and metabolism impairment. Consequently, they are not suggested in the treatment of TBI in animals, either.14
Hyperglycemia exacerbates secondary brain injury and it is index of the severity of trauma to neural tissue, especially during the first 24 hours.51 Furthermore, in humans it has been associated with bad prognosis because negative outcome was observed in cases of serum glucose levels above 200 mg/dl.27 The same conclusion was reached in an animal study in which TBI was experimentally induced.28 This is another reason -apart from that associated with the osmotic effects- which explains why dextrose 5 % solution is not used in the treatment of TBI. Iatrogenic hyperglycemia (e.g. corticosteroid administration) must always be avoided4 while in people closed monitoring of glucose levels is achieved by continuing measurements and judicious use of insulin.17
Hypothermia has been used in people52,53 and recently in dogs with TBI54 and it may decrease the metabolic needs of the brain, cerebral edema and IP. Clinical efficacy of hypothermia is questioned, but hyperthermia is certainly contraindicated.8
3. Other measures
- Analgesia, usually by using opioids.55
- Elevation of the animal’s head at a 30º angle, by placing the animal on an inclined surface.56
- Barbiturate coma, recruited only in cases refractory to other treatments because it causes hypotension and hypoventilation which may have detrimental effect to the animal.8
- Administration of H2 blockers or proton pump inhibitors and sucralfate for the prophylaxis of gastric ulcer which occurs in cases of hemorrhage from the gastrointestinal tract, in patients with TBI.57
- Enteral feeding and simultaneous treatment with procinetic drugs plus placement of nasoesophageal tube. In patients with loss of consciousness or absent pharyngeal reflex parenteral nutrition is recommended.8
> TBI prognosis
Patients suffering from TBI may develop various complications like haemostatic disorders, pneumonia, sepsis, temporary or permanent diabetes insipidus and seizures.58 Seizures may appear months to years after the TBI.1 In people, seizures are reported to occur at a rate of 4-42 % in cases of severe TBI39 but in veterinary patients this rate is unknown and it is assumed to be small.
Treatment of animals with TBI must be immediate and aggressive so that they will survive and be acceptable as pets by their owners. The aim of treatment in TIB is to give animals a good quality of life. Many of them do rehabilitate if systematic and neurological injuries are recognized and treated early. As it was previously mentioned, dogs and cats show considerable rehabilitation
ability despite the loss of neural tissue. It is therefore important not to infer hasty conclusions about prognosis based on the initial status of the animal.8
In people, prognostic factors which determine the outcome in TBI are considered the age, etiology, scoring based on Glasgow coma scale, pupil response to light, computed tomography findings, occurrence of hypotension and hypoxia, glucose concentration and prothrombin time. In small animal practice it is not known which parameters influence prognosis of TBI but recently scoring based on MGCS was associated with survival in dogs during the first 48 hours. For example if grade 8 based on MGCS is assigned to a dog, then a 50 % survival rate is anticipated. In the same study sex, weight, age and skull fractures did not seem to have any effect on survival, albeit patients with systematic disorders were excluded. 21 However, in patients with multiple injuries, prognostic significance of scoring based on MGCS must be interpreted with caution.8 Therefore, effectiveness of treatment and prognosis in patients with TBI will be always hard to assess due to the multifactorial nature of the injuries.
> References
1. Dewey CW. Emergency management of the head trauma patient. Principles and practice. Vet Clin North Am Small Anim Pract. 2000, 30: 207-225.
2. Hopkins AL. Head trauma. Vet Clin North Am Small Anim Pract. 1996, 26: 875-891.
3. Shores A. Craniocerebral Trauma. In: Kirk’s Current Veterinary Therapy. Bonagura JD, Kirk RW, Twedt DC (ed). 10th edn. Elsevier, Saunders: Philadelphia, 1989, p. 847-853.
4. Syring RS, Otto CM, Drobatz KJ. Hyperglycemia in dogs and cats with head trauma: 122 cases (1997-1999). J Am Vet Med Assoc. 2001, 218: 1124-1129.
5. Fletcher DJ. Head trauma management. In: Congress proceedings of the IVECCS. New Orleans, U.S.A., 2007, pp. 403-410.
6. Dewey C, Downs M, Aron D. Acute traumatic intracranial haemorrhage in dogs and cats. A retrospective evaluation of 23 cases. Vet Comp Orthop Traumatol. 1993, 6: 153-8.
7. Platt SR, Radaelli ST, McDonnell JJ. Computed tomography after mild head trauma in dogs. Vet Rec. 2002, 151: 243.
8. Sande A, West C. Traumatic brain injury: a review of pathophysiology and management. J Vet Emerg Crit Care (San Antonio). 2010, 20: 177-190.
9. Chesnut RM. The management of severe traumatic brain injury. Emerg Med Clin North Am. 1997, 15: 581-604.
10. 1Sturges B, LeCouteur RA. Intracranial Hypertension. In: Small Animal Critical Care Medicine. Silverstein DC, Hopper K (ed). 1st edn. Saunders Elsevier: Missouri, 2009, pp. 423-429.
11. Raisis AL, Brearley JC. Anaesthesia, analgesia and supportive care. In: Bsava Manual Of Canine and Feline Neurology. Platt SR, Platt S, Olby NJ (ed). 3rd edn. British Small Animal Veterinary Association: Gloucester 2004, pp. 337-354.
12. Zink BJ. Traumatic brain injury. Emerg Med Clin North Am. 1996, 14: 115-150.
13. Bouma GJ, Muizelaar JP, Bandoh K, Marmarou A. Blood pressure and intracranial pressure-volume dynamics in severe head injury: relationship with cerebral blood flow. J Neurosurg. 1992, 77: 15-19.
14. Syring RS. Assessment and treatment of central nervous system abnormalities in the emergency patient. Vet Clin North Am Small Anim Pract. 2005, 35: 343-358.
15. Guyton AC, Hall JE. Nervous regulation of the circulation, and rapid control of arterial pressure. In: Guyton and Hall Textbook of Medical Physiology. Guyton AC, Hall JE (ed). 11th edn. Elsevier Health Sciences: Philadelphia, 2006, pp. 212-213.
16. Chesnut RM, Marshall LF, Klauber MR, Blunt BA, Baldwin N, Eisenberg HM, Jane JA, Marmarou A, Foulkes MA. The role of secondary brain injury in determining outcome from severe head injury. J Trauma. 1993, 34: 216-222.
17. Rostami E, Bellander BM. Monitoring of glucose in brain, adipose tissue, and peripheral blood in patients with traumatic brain injury: a microdialysis study. J Diabetes Sci Technol. 2011, 5: 596-604.
18. Vespa P, Bergsneider M, Hattori N, Wu HM, Huang SC, Martin NA, Glenn TC, McArthur DL, Hovda DA. Metabolic crisis without brain ischemia is common after traumatic brain injury: a combined microdialysis and positron emission tomography study. J Cereb Blood Flow Metab. 2005, 25: 763-774.
19. Rosner MJ, Newsome HH, Becker DP. Mechanical brain injury: the sympathoadrenal response. J Neurosurg. 1984, 61: 76-86.
20. Siegel JH. The effect of associated injuries, blood loss, and oxygen debt on death and disability in blunt traumatic brain injury: the need for early physiologic predictors of severity. J Neurotrauma. 1995, 12: 579-590.
21. Platt SR, Radaelli ST, McDonnell JJ. The prognostic value of the modified Glasgow Coma Scale in head trauma in dogs. J Vet Intern Med. 2001, 15: 581-584.
22. Maas AI, Dearden M, Teasdale GM, Braakman R, Cohadon F, Iannotti F, Karimi A, Lapierre F, Murray G, Ohman J, Persson L, Servadei F, Stocchetti N, Unterberg A. EBIC-guidelines for management of severe head injury in adults. European Brain Injury Consortium. Acta Neurochir (Wien). 1997, 139: 286-294.
23. Gruen P, Liu C. Current trends in the management of head injury. Emerg Med Clin North Am. 1998, 16: 63-83.
24. Bush WW. Pathophysiology of the head injured patient. In: Congress proceedings of the IVECCS. San Antonio, U.S.A., 2006, pp. 481-485.
25. Scherer MR, Burrows H, Pinto R, Littlefield P, French LM, Tarbett AK, Schubert MC. Evidence of central and peripheral vestibular pathology in blast-related traumatic brain injury. Otol Neurotol. 2011, 32: 571-580.
26. Drobatz KJ, Saunders HM, Pugh CR, Hendricks JC. Noncardiogenic pulmonary edema in dogs and cats: 26 cases (1987-1993). J Am Vet Med Assoc. 1995, 206: 1732- 1736.
27. Rovlias A, Kotsou S. The influence of hyperglycemia on neurological outcome in patients with severe head injury. Neurosurgery. 2000, 46: 335-342.
28. Cherian L, Hannay HJ, Vagner G, Goodman JC, Contant CF, Robertson CS. Hyperglycemia increases neurological damage and behavioral deficits from posttraumatic secondary ischemic insults. J Neurotrauma. 1998, 15: 307-321.
29. Clifton GL, Miller ER, Choi SC, Levin HS. Fluid thresholds and outcome from severe brain injury. Crit Care Med. 2002, 30: 739-745.
30. Zhuang J, Shackford SR, Schmoker JD, Pietropaoli JA, Jr. Colloid infusion after brain injury: effect on intracranial pressure, cerebral blood flow, and oxygen delivery. Crit Care Med. 1995, 23: 140-148.
31. Kaieda R, Todd MM, Cook LN, Warner DS. Acute effects of changing plasma osmolality and colloid oncotic pressure on the formation of brain edema after cryogenic injury. Neurosurgery. 1989, 24: 671-678.
32. Wisner D, Busche F, Sturm J, Gaab M, Meyer H. Traumatic shock and head injury: effects of fluid resuscitation on the brain. J Surg Res. 1989, 46: 49-59.
33. Boag A, Hughes D. Fluid Therapy. In: BSAVA Manual Of Canine and Feline Emergency and Critical Care. King L, Hammond R (ed). British Small Animal Veterinary Association: Gloucester, 2001, pp. 36-37.
34. Kreimeier U, Messmer K. Small-volume resuscitation: from experimental evidence to clinical routine. Advantages and disadvantages of hypertonic solutions. Acta Anaesthesiol Scand. 2002, 46: 625-638.
35. Prough DS, Whitley JM, Taylor CL, Deal DD, DeWitt DS. Regional cerebral blood flow following resuscitation from hemorrhagic shock with hypertonic saline. Influence of a subdural mass. Anesthesiology. 1991, 75: 319-327.
36. Smith GJ, Kramer GC, Perron P, Nakayama S, Gunther RA, Holcroft JW. A comparison of several hypertonic solutions for resuscitation of bled sheep. J Surg Res. 1985, 39: 517-28.
37. McGraw CP, Howard G. Effect of mannitol on increased intracranial pressure. Neurosurgery. 1983, 13: 269-271.
38. Muizelaar JP, Lutz HA, 3rd, Becker DP. Effect of mannitol on ICP and CBF and correlation with pressure autoregulation in severely head-injured patients. J Neurosurg. 1984, 61: 700-706.
39. Bratton SL, Chestnut RM, Ghajar J, McConnell Hammond FF, Harris OA, Hartl R, Manley GT, Nemecek A, Newell DW, Rosenthal G, Schouten J, Shutter L, Timmons SD, Ullman JS, Videtta W, Wilberger JE, Wright DW. Guidelines for the management of severe traumatic brain injury. II. Hyperosmolar therapy. J Neurotrauma. 2007, 24 Suppl 1: S14-20.
40. Marshall LF, RW SM, Rauscher LA, Shapiro HM. Mannitol dose requirements in brain-injured patients. J Neurosurg. 1978, 48: 169-172.
41. Barry KG, Berman AR. Mannitol infusion. III. The acute effect of the intravenous infusion of mannitol on blood and plasma volumes. N Engl J Med. 1961, 264: 1085-1058.
42. Mizoi K, Suzuki J, Imaizumi S, Yoshimoto T. Development of new cerebral protective agents: the free radical scavengers. Neurol Res. 1986, 8: 75-80.
43. Zornow MH, Todd MM, Moore SS. The acute cerebral effects of changes in plasma osmolality and oncotic pressure. Anesthesiology. 1987, 67: 936-941.
44. Qureshi AI, Suarez JI, Bhardwaj A, Mirski M, Schnitzer MS, Hanley DF, Ulatowski JA. Use of hypertonic (3%) saline/acetate infusion in the treatment of cerebral edema: Effect on intracranial pressure and lateral displacement of the brain. Crit Care Med. 1998, 26: 440- 446.
45. Qureshi AI, Wilson DA, Traystman RJ. Treatment of elevated intracranial pressure in experimental intracerebral hemorrhage: comparison between mannitol and hypertonic saline. Neurosurgery. 1999, 44: 1055-1063.
46. Kerwin AJ, Schinco MA, Tepas JJ, 3rd, Renfro WH, Vitarbo EA, Muehlberger M. The use of 23.4% hypertonic saline for the management of elevated intracranial pressure in patients with severe traumatic brain injury: a pilot study. J Trauma. 2009, 67: 277-282.
47. Coles JP, Minhas PS, Fryer TD, Smielewski P, Aigbirihio F, Donovan T, Downey SP, Williams G, Chatfield D, Matthews JC, Gupta AK, Carpenter TA, Clark JC, Pickard JD, Menon DK. Effect of hyperventilation on cerebral blood flow in traumatic head injury: clinical relevance and monitoring correlates. Crit Care Med. 2002, 30: 1950-1959.
48. Roberts I, Yates D, Sandercock P, Farrell B, Wasserberg J, Lomas G, Cottingham R, Svoboda P, Brayley N, Mazairac G, Laloe V, Munoz-Sanchez A, Arango M, Hartzenberg B, Khamis H, Yutthakasemsunt S, Komolafe E, Olldashi F, Yadav Y, Murillo-Cabezas F, Shakur H, Edwards P. Effect of intravenous corticosteroids on death within 14 days in 10008 adults with clinically significant head injury (MRC CRASH trial): randomised placebo-controlled trial. Lancet. 2004, 364: 1321-1328.
49. Edwards P, Arango M, Balica L, Cottingham R, El- Sayed H, Farrell B, Fernandes J, Gogichaisvili T, Golden N, Hartzenberg B, Husain M, Ulloa MI, Jerbi Z, Khamis H, Komolafe E, Laloe V, Lomas G, Ludwig S, Mazairac G, Munoz Sanchez Mde L, Nasi L, Olldashi F, Plunkett P, Roberts I, Sandercock P, Shakur H, Soler C, Stocker R, Svoboda P, Trenkler S, Venkataramana NK, Wasserberg J, Yates D, Yutthakasemsunt S. Final results of MRC CRASH, a randomised placebo-controlled trial of intravenous corticosteroid in adults with head injury-outcomes at 6 months. Lancet. 2005, 365: 1957-1959.
50. Lei J, Gao GY, Jiang JY. Is management of acute traumatic brain injury effective? A literature review of published Cochrane Systematic Reviews. Chin J Traumatol. 2012, 15: 17-22.
51. Liu-DeRyke X, Collingridge DS, Orme J, Roller D, Zurasky J, Rhoney DH. Clinical impact of early hyperglycemia during acute phase of traumatic brain injury. Neurocrit Care. 2009, 11: 151-157.
52. Marion DW, Penrod LE, Kelsey SF, Obrist WD, Kochanek PM, Palmer AM, Wisniewski SR, DeKosky ST. Treatment of traumatic brain injury with moderate hypothermia. N Engl J Med. 1997, 336: 540-546.
53. Clifton GL, Miller ER, Choi SC, Levin HS, McCauley S, Smith KR, Jr., Muizelaar JP, Wagner FC, Jr., Marion DW, Luerssen TG, Chesnut RM, Schwartz M. Lack of effect of induction of hypothermia after acute brain injury. N Engl J Med. 2001, 344: 556-563.
54. Hayes GM. Severe seizures associated with traumatic brain injury managed by controlled hypothermia, pharmacologic coma, and mechanical ventilation in a dog. J Vet Emerg Crit Care (San Antonio). 2009, 19: 629-634.
55. Lauer KK, Connolly LA, Schmeling WT. Opioid sedation does not alter intracranial pressure in head injured patients. Can J Anaesth. 1997, 44: 929-933.
56. Ng I, Lim J, Wong HB. Effects of head posture on cerebral hemodynamics: its influences on intracranial pressure, cerebral perfusion pressure, and cerebral oxygenation. Neurosurgery. 2004 Mar, 54: 593-597; discussion 8.
57. Simons RK, Hoyt DB, Winchell RJ, Holbrook T, Eastman AB. A risk analysis of stress ulceration after trauma. J Trauma. 1995, 39: 289-293.
58. Piek J, Chesnut RM, Marshall LF, van Berkum- Clark M, Klauber MR, Blunt BA, Eisenberg HM, Jane JA, Marmarou A, Foulkes MA. Extracranial complications of severe head injury. J Neurosurg. 1992, 77: 901-907.
